
This page is intended for UK healthcare professionals only.
This non-promotional content is organised and funded by CarbonNil Medical.
About dRTA
A non-promotional, educational presentation on Distal Renal Tubular Acidosis
Basis of Renal Physiology1,2
- The nephron is the functional unit of kidneys (400,000 to 800,000 nephrons per kidney)
- Each nephron is made of 1 glomerulus and 1 renal tubule, in continuity.
- Each kidney tubule has four different parts, and each part performs a specific function. As the filtered fluid flows through them, substances are added to or removed from it by exchanging materials with the blood, gradually turning it into urine.
- At the end of the tubule, urine composition is final.
- Along the tubule, exchanges are controlled by hormones and mediators either of systemic or local origin.
Urine formation / Proximal Convoluted Tubule/ Tubulopathy1-5
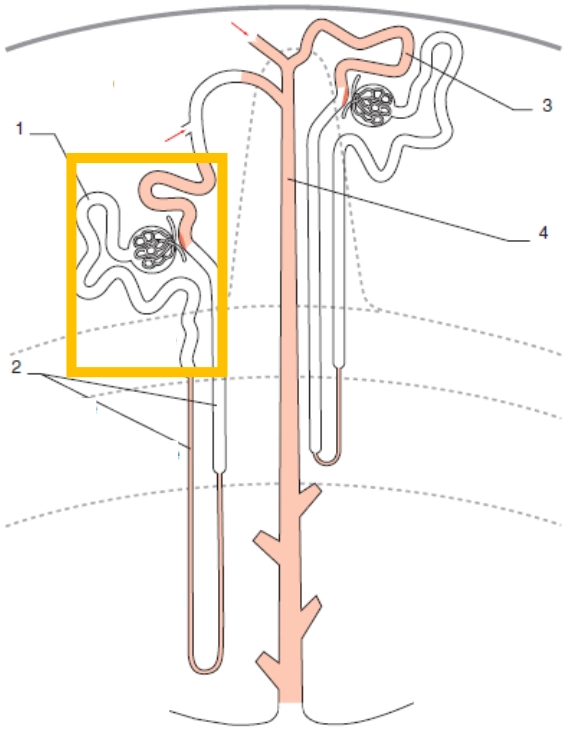
1. Proximal Tubule
2. Loop of Henle
3. Distal convoluted tubule
4. Collecting duct
Major reabsorption segment:
- 100% of glucose and amino acids
- 60 to 70% of water, sodium, phosphates
- 80% of bicarbonates
Partial or total alteration of proximal convoluted tubule functions:
- Primary reno-tubular Fanconi syndrome (rare genetic renal disease): high loss of glucose, bicarbonate, phosphate, uric acid, potassium, sodium and some amino-acids)
- Secondary Fanconi syndrome:
- Genetic diseases: cystinosis, Dent disease, Lowe syndrome
- Acquired causes: drugs or heavy metal poisoning, malignancies
= Proximal RTA type 2 (metabolic acidosis with hypokalemia)
Urine formation / Henle’s Loop/ Tubulopathy1-3
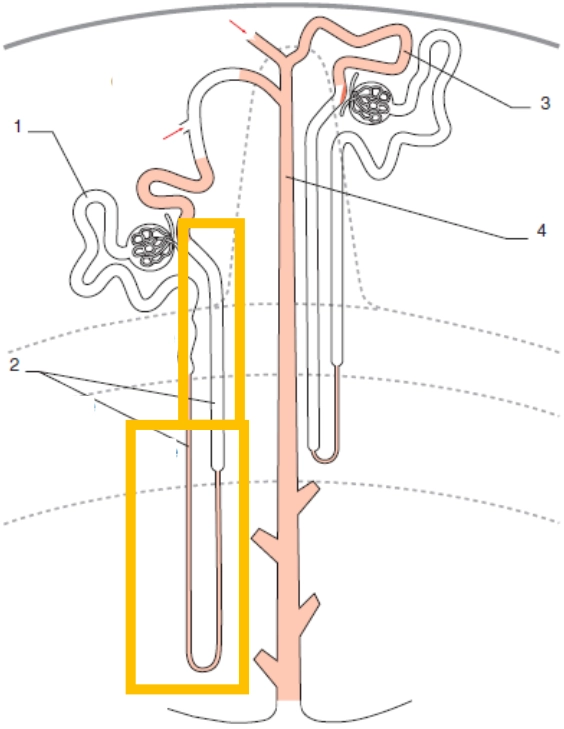
1. Proximal Tubule
2. Loop of Henle
3. Distal convoluted tubule
4. Collecting duct
Reabsorption segment (completes proximal segment):
- 20 to 30% of sodium
- 10 to15% of bicarbonates
- 30% of calcium
- 70% of magnesium
Alteration of exchanges in this tubular segment: Bartter syndrome (major hydro electrolytic loss): metabolic alcalosis with hypokalemia
Urine formation / Distal Tubule (2 sub segments) / Tubulopathies1-3
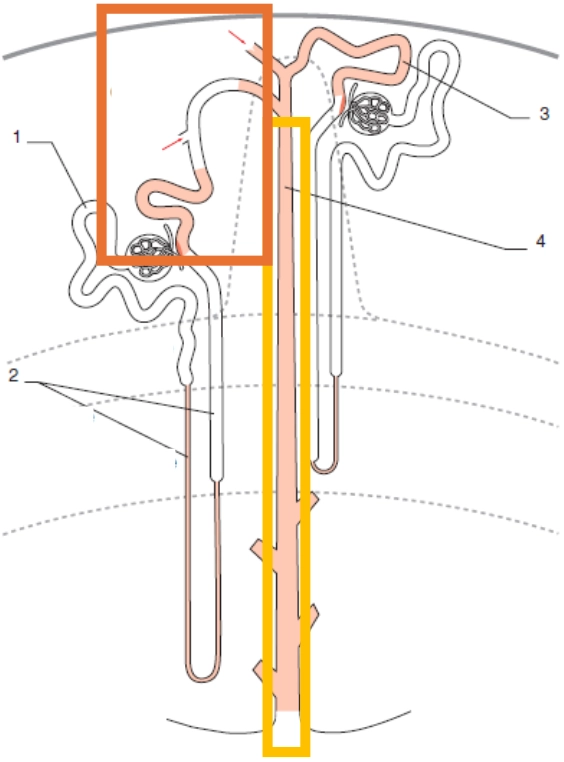
1. Proximal Tubule
2. Loop of Henle
3. Distal convoluted tubule
4. Collecting duct
Distal convoluted tubule:
- Low reabsorption segment in terms of quantity (less than 10% of the filtrated volume)
- Very important exchange step for qualitative hydro-electrolytic adjustment
- An alteration of exchanges in this sub segment: Gitelman syndrome (metabolic alkalosis with hypokalaemia)
Collecting duct:
- Major site of potassium/ acids secretion and sodium/water reabsorption
- In the event of acid secretion failure, due to proton pump alteration: distal RTA type 1 or Albright acidosis or « classical » RTA (metabolic acidosis with hypokalemia)
- In the event of NH4+ production mostly due to hypoaldosteronism: distal RTA type 4 (metabolic acidosis with hyperkalaemia)
Regulation of acid-base balance6,7
- pH (power of hydrogen) represents the concentration of hydrogen ions (H⁺) in a solution, which determines its acidity. Under normal physiological conditions, the concentration of H⁺ in blood is extremely low.
- The human body needs to maintain this low acid quantity despite food/ beverage intake (animal proteins bring the highest quantity of acids).
- To do so, the main buffering system is carbonic acid / bicarbonate:
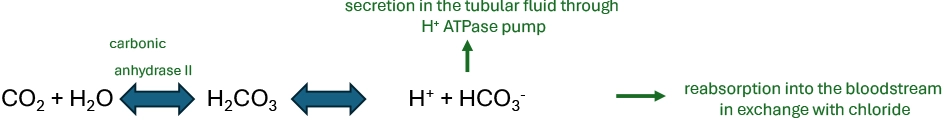
- pH changes when the ratio between PCO2 and HCO3- is modified
- if PCO2 changes: respiratory acidosis (PCO2 increase) or alkalosis (PCO2 decrease)
- if HCO3- changes: metabolic acidosis (HCO3- decrease) or alkalosis (HCO3- increase)
- One of the function of the lungs is CO2 excretion
- Hypoventilation=respiratory acidosis
- Hyperventilation= respiratory alkalosis
- A decrease in blood pH leads to hyperventilation and conversely, an increase in blood pH in blood pH leads to hypoventilation
- One of the functions of the kidneys is filtrated bicarbonate reabsorption (proximal tubule) and acid excretion (distal tubule)
Acid-base disorders: metabolic acidosis diagnosis6
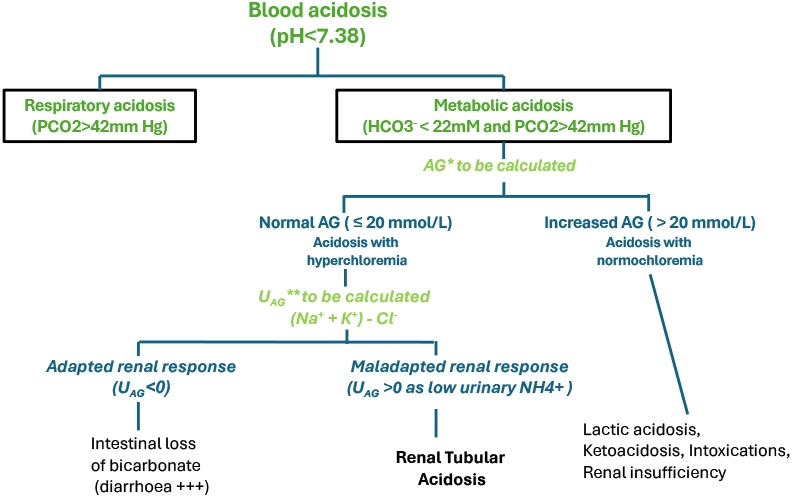
*AG: Plasma anion gap
**UAG: Urine anion gap
Acid-base balance: Plasma anion gap (AG)6
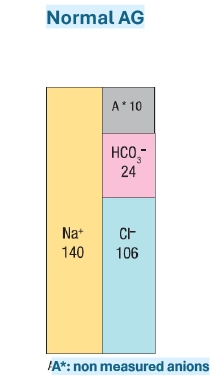
- Due to plasma electro-neutrality, cations concentration is equal to anions concentration.
- When specifically considering Na+ et K+ cations and HCO3- et Cl- anions, their plasma concentration difference is called anion gap.
Normal plasma anion gap =16±4 mmole/L
- In practice, there are physiologically more non measured anions than non measured cations.
- AG changes will allow to determine if the anion compensating the excess of H+ (bicarbonate loss) is Cl- or anion which is non measured .
Acid-base disorders: metabolic acidosis6
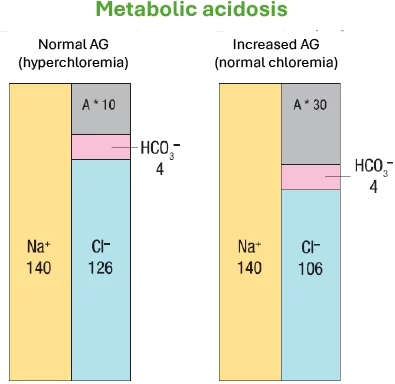
A*: Non measured anion
Metabolic acidosis with normal AG (with hyperchloremia):
Intestinal or renal losses of HCO3-.which is compensated by a proportional increase in Cl- (HCl gain) leading to a metabolic acidosis with hyperchloremia.
Metabolic acidosis with increased AG ( H+ associated to a non measured anion, i.e. lactate) (with normal chloremia)
Presence of another acid than HCl and AG increase as HCO3- decrease is compensated by a non measured anion leading to a metabolic acidosis with normal chloremia.
Acid-base disorders: metabolic acidosis, UAG6
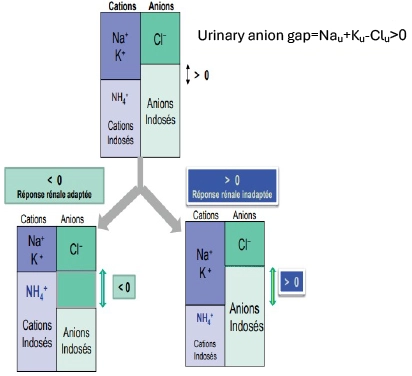
When AG is normal, renal response can be assessed by urinary anion gap (UAG) which is inversely proportional to ammoniuria (NH4+ urinary concentration)
UAG < 0 (= high urinary NH4+ concentration = adapted renal response) => extrarenal origin of metabolic acidosis (bicarbonate intestinal loss).
UAG > 0 (= low urinary NH4+ concentration = non adapted renal response) => tubular renal origin of metabolic acidosis
Renal Tubular
Acidosis1-2,7,8
Four types of renal tubular acidosis:
- Type 1: Distal renal tubular acidosis (dRTA) with hypokalaemia due to H+ secretion default in the presence of metabolic acidosis. Alkaline load test: UpH>5.5 and maladapted renal response to acid load test as UpH still >5.5
- Type 2: Proximal renal tubular acidosis with hypokalemia due to HCO3- reabsorption default
- Alkaline load test: UpH<5.5
- Type 3: Distal and proximal renal tubular acidosis with hypokalemia (congenital dysfunction of carbonic anhydrase located in cells of proximal tubule, Henle’s loop and collecting tubule)
- Type 4: Distal renal tubular acidosis with hyperkalemia due to NH4+ production default secondary to aldosterone deficiency or renal tubular resistance to aldosteron
Type 1 distal Renal Tubular Acidosis or dRTA7-9
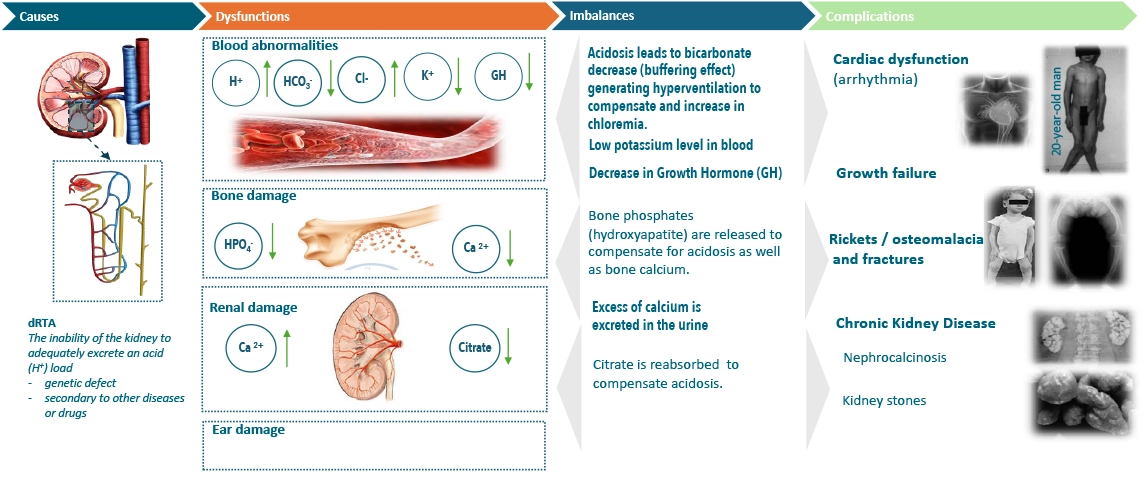
- dRTA is a rare disease where the distal tubule is unable to secrete protons (H+) in urine in response to a metabolic acidosis, thus leading to an impairment of the acid-base homeostasis.
- dRTA is due to pathogenic variants in genes involved in acid excretion leading to modification of transporters or transcription factors implicated in the renal protons’ secretion. These variants can be either genetic (primary or inherited dRTA) or affected as a consequence of an injury, disease or drug intake, inducing nephrotoxicity leading to tubule damage (secondary or acquired dRTA).
Type 1 dRTA UK prevalence10
- Retrospective analysis showed that dRTA prevalence in 2017 was estimated to be between 0.46 (recorded cases, of which 22.1% were considered primary) and 1.60 when including suspected cases (7.6% primary) per 10,000 people.
- Prescription and clinical records of diagnosed patients revealed a wide range of comorbidities and a need for pharmacological treatment to manage associated symptoms.
- The study provides new estimates of dRTA prevalence in Europe and suggests that patients may often be unreported or miscoded, potentially confounding appropriate disease management.
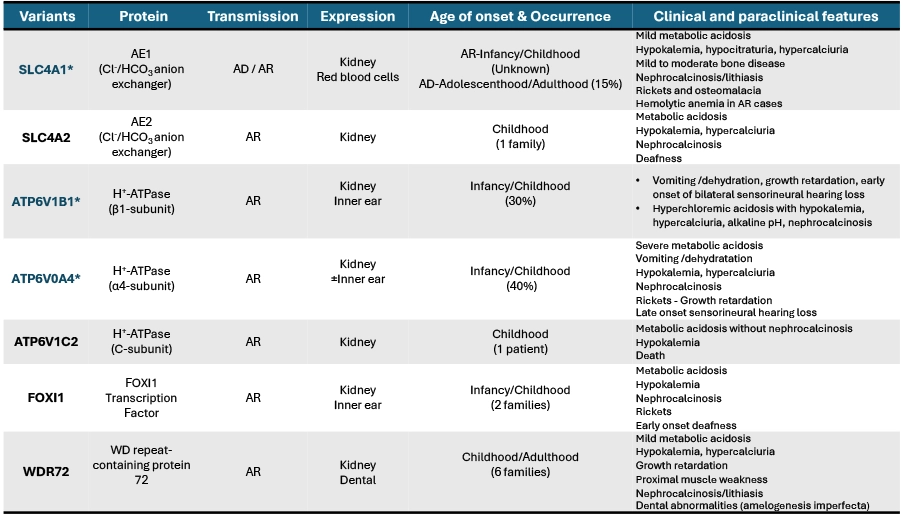
Inherited type 1 dRTA7,8,11,12
- Inherited tubulopathies are secondary to pathogenic mutations in gene coding either for specific transporters of solutes or for their regulating proteins. They are classified according to the concerned solute or segment.
- Inherited dRTA occurs mostly in children, and is diagnosed from the 1st year of life depending upon the mutation
- Primary dRTA is the most frequent form of primary RTA in western countries
- Several gene variants are responsible, and their transmission can be autosomal recessive (AR) or autosomal dominant (AD)
- Analysis of the genetic defects involved is important to define functional consequences and possible genotype-phenotype correlation
Inherited type 1 dRTA7,8
- Homozygous, biallelic presentation of:
-ATP6V0A4
-ATP6V1B1
-ATP6V1C2
-FOXI1
-WDR72
-SLC4A2 - Homozygous, biallelic presentation of:
-SLC4A1
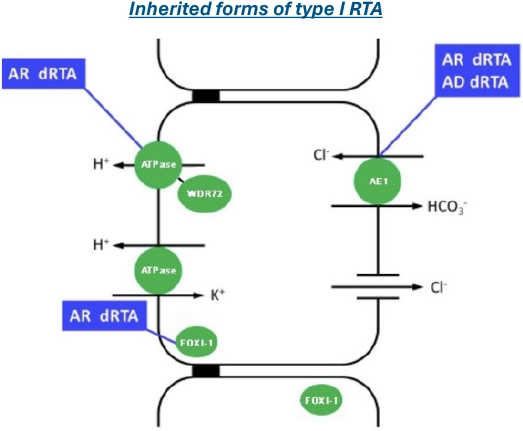
Inherited dRTA: genotype / phenotype correlation7,8,13

Acquired type 1 dRTA7,8
- Diagnosed mostly in adulthood
- Associated with:
A systemic disease
-
- Auto-immune diseases: Sjögren syndrome (SS), systemic lupus erythematosus (SLE), amyloidosis, thyroiditis, primary biliary cirrhosis, auto-immune hepatitis.
Nephrocalcinosis
-
- Medullary sponge kidney (MSK) / Cacchi Ricci disease
- Hyperparathyroidism
Drugs
-
- Toluene, cyclamate
Acquired dRTA / Sjögren Syndrome14
- Auto immune, chronic disease with lymphocyto-plasmocytic infiltration of exocrine glands (predominantly the salivary and lacrimal glands) leading to sicca symptoms.
- Mean age of Sjögren patients: 50 years with majority of female patients (ratio 9/1)
- Extra-glandular disease involving multiple organs may occur, including kidney disease
- Renal complications, in less of 10% of Sjögren patients, are mainly tubulo-interstitial nephritis (due to lymphocyto-plasmocytic infiltration) but also dRTA, diabetes insipidus, Fanconi syndrome…
dRTA - Main clinical features13
- Potential presentation clinical symptoms
- Infants: failure to thrive, growth retardation, polyuria, polydipsia, vomiting, constipation, rickets, hypotonia, nephrocalcinosis, haemolytic anaemia, sensorineural hearing loss
- Children and adolescents: growth retardation, polyuria, polydipsia, rickets, vomiting, constipation, muscle weakness/hypokalaemic paralysis, haemolytic anaemia, sensorineural hearing loss, nephrocalcinosis/urolithiasis, enamel defects
- Adults: nephrocalcinosis/urolithiasis, osteomalacia, muscle weakness/hypokalaemia, bone pain, fractures, haemolytic anaemia, sensorineural hearing loss, enamel defects
dRTA - Main biochemistry
features6-8,13
- Metabolic acidosis (arterial blood pH < 7.38*)
- Low serum bicarbonate for buffering effect (< 22 mmol/l) inducing a hyperventilation to compensate
- Normal plasma anion gap due to compensation of bicarb decrease by hyperchloremia
- Hypokalaemia (< 3.5 mmol/l)
- Impaired urine acidification (inappropriate elevated urine pH ( > 5.5), due to ineffective renal response to blood acidosis)
- Positive urine anion gap (hypoammoniuria=tubular origin of metabolic acidosis)
- Hypocitraturia as plasma acidosis enhances citrate proximal tubular reabsorption
- Hypercalciuria due to buffering of plasma acidosis with bone hydroxyapatite with concomitant increase in serum calcium
- Decrease in GH secretion due to acidosis and induced resistance to GH
*Normal venous blood pH: 7.32-7.38; metabolic acidosis when venous blood pH < 7.32
References
- CUEN, Ellipse Edition.2022: Chapt 1
- Albert Z. J. Interven. Nephro. 2022; 5(5):66–69
- Blanchard A et al. Nephrol & Therap.2009; 5: 68-83.2009.
- Orphanet cystinosis https://www.orpha.net/en/disease/detail/213. [accessed January 2026]
- Orphanet Primary Fanconi reno-tubular syndrome https://www.orpha.net/en/disease/detail/3337 [accessed January 2026]
- CUEN, Ellipse Edition.2022: Chapt 5.
- Giglio S et al. J of Nephrology, 2021,34:2073-2083
- Reddi AS. 2020, Springer Chapt 8.
- Lopez-Garcia S C et al., Nephrol Dial Transplant. 2019,34:981-991
- Bianic F. Et al.Nephron 2021;145(5):486-495 https://pubmed.ncbi.nlm.nih.gov/34198293/
- Boyer O et al.2022, J of Nephrol. 35:2119–2122
- Santos F et al. Ped Nephrol.2015.30: 2099-2017.
- Trepiccione F et al. 2021.Nephrol Dial Transplant;36:1585-1596
- Mariette X, François H. Néphrologie & Thérapeutique, 2020, 16 (7), pp.440-452
dRTA in summary7,8,13
NP/CNM/2026/020 January 2026